
Análisis de la guía General Wellness Policy for Low Risk Devices (FDA, 2026)
Por Maria Isabel Iñigo Petralanda (*)

El 6 de enero de 2026 la Agencia Estadounidense Food and Drug Administration (FDA) publicó la actualización de la guía General Wellness: Policy for Low Risk Devices https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-wellness-policy-low-risk-devices, cuyo objetivo es clarificar los criterios bajo los cuales determinados productos orientados al bienestar general (general wellness products) pueden quedar fuera del alcance de la regulación aplicable a los dispositivos médicos (medical devices). La guía, -emitida por el Center for Devices and Radiological Health (CDRH) con la participación del Digital Health Center of Excellence (oficina creada por la FDA para coordinar las líneas regulatorias vinculadas a software, inteligencia artificial (IA) y dispositivos digitales (2)-, establece que la calificación que otorga la agencia, depende de dos factores centrales: el uso previsto (intended use) y el nivel de riesgo asociado. Entre “las novedades” de esta Guia, se destaca la posibilidad de que ciertos dispositivos con sensores fisiológicos —como wearables que estiman parámetros de salud— permanezcan fuera del ámbito sanitario regulado siempre que no realicen funciones diagnósticas, terapéuticas o de manejo clínico. Este artículo analiza la naturaleza jurídica, el contexto regulatorio y el impacto potencial de la Guía en el desarrollo de soluciones de bienestar digital, así como su posible influencia en otras jurisdicciones fuera de los Estados Unidos. Y discute las implicancias para el ecosistema latinoamericano, especialmente en relación con procesos de digitalización del cuidado y la telemedicina.
Mercado, tecnología y regulación. El mercado global de bienestar digital ha experimentado una expansión significativa en los últimos años, impulsado principalmente por el desarrollo de dispositivos portables, aplicaciones móviles y plataformas de auto-seguimiento fisiológico vinculadas a estilos de vida saludables, deporte, sueño y nutrición. La convergencia entre capacidades de sensorización fisiológica y usos no clínicos generó dudas regulatorias sobre el punto en que una tecnología orientada al bienestar general debe ser considerada un dispositivo médico y, por lo tanto, quedar sujeta a requisitos regulatorios específicos. La actualización de la FDA publicada en 2026 y que esta Guia bajo análisis documenta, ofrece una delimitación operativa de esta frontera.
La Guía establece desde su prefacio un documento de recomendaciones no vinculantes (“nonbinding recommendations”), característica típica en la tradición regulatoria estadounidense para documentos de interpretación y política de cumplimiento (enforcement policy). Es así que, este tipo de instrumentos no crea obligaciones legales nuevas, sino que explicita criterios que la agencia aplicará al evaluar el cumplimiento del marco normativo vigente (1). Este punto es bastante conclusivo en términos de fuerza regulatoria.
Criterios regulatorios: intended use y nivel de riesgo. Para comprender la naturaleza y alance del documento es importante recordar que esta Guía 2026 reemplaza la versión de septiembre de 2019 e incorpora elementos derivados de la implementación del 21st Century Cures Act https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act , particularmente la exclusión de ciertos software orientados al mantenimiento de estilos de vida saludables del alcance de la definición legal de dispositivo (3).
Si algo es pedido a una agencia de calidad como la FDA, es definir claramente los ejes de análisis regulatorios. En el caso de esta Guia 2026 son dos criterios taxonómicos: el uso previsto y el nivel de riesgo.
- Uso previsto (intended use). Quedan contemplados los productos de bienestar general aquellos que: promueven estados generales de salud (p. ej., manejo del estrés, sueño, peso, actividad física), o promueven estilos de vida saludables que pueden contribuir a reducir el riesgo o permitir una mejor convivencia con enfermedades crónicas, siempre sin finalidad diagnóstica, terapéutica o de manejo clínico (1,4).
- Nivel de riesgo. La clasificación como producto de bienestar requiere asimismo que el dispositivo: a) no sea invasivo; b) no sea implantable; c) no utilice tecnologías que impliquen riesgos sin controles regulatorios (p. ej., láseres, neuroestimulación), y d) no sustituya o interfiera con dispositivos aprobados con finalidad clínica (1).
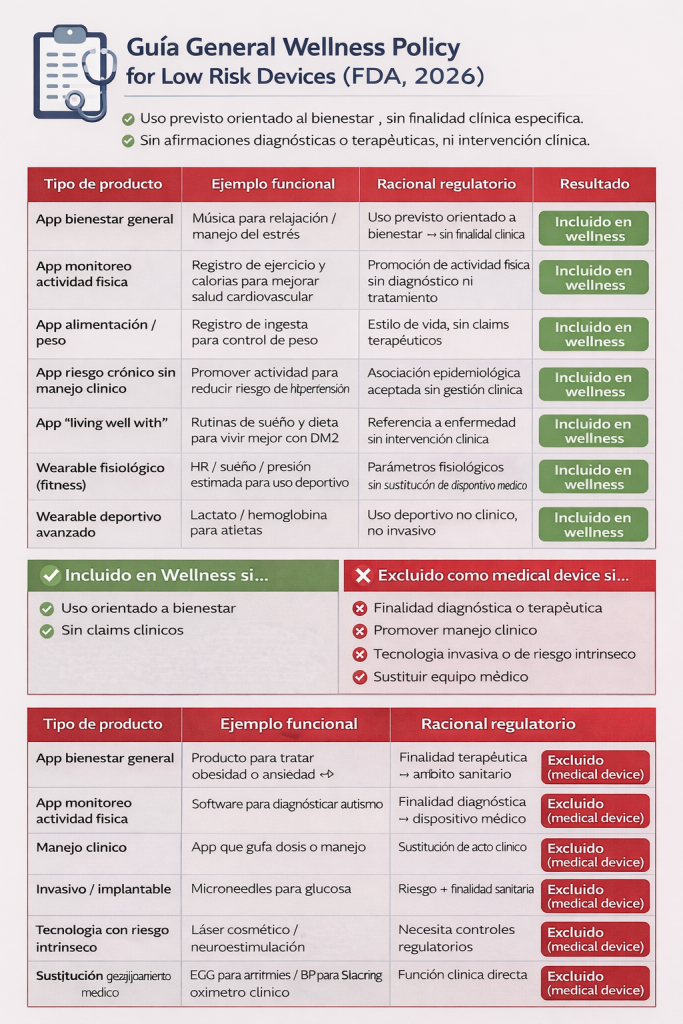
Wearables y sensorización fisiológica: la novedad 2026. El aspecto más disruptivo de la actualización es la inclusión de wearables capaces de estimar o inferir parámetros biométricos —por ejemplo, estimación no clínica de presión arterial, variabilidad de la frecuencia cardíaca, saturación de oxígeno o lactato— siempre que dichas funciones carezcan de finalidad diagnóstica, terapéutica o de manejo clínico.
Esta interpretación se aparta de la lógica tradicional, según la cual la sola medición de un parámetro fisiológico activaba el ámbito regulatorio sanitario. El cambio implica que la función clínica depende de la finalidad y no del sensor.
Bajo la nueva interpretación, quedarán excluidos de la guía los productos que: a. emitan alertas clínicas; b. utilicen umbrales diagnósticos; c. recomienden acciones terapéuticas; d. sustituyan equipos médicos o realicen screening, diagnóstico, monitorización o manejo clínico (1,5).
Contexto internacional y comparación regulatoria. Estados Unidos no es la única jurisdicción con procesos de clarificación normativa. En la Unión Europea, el Reglamento MDR https://eur-lex.europa.eu/legal-content/ES/ALL/?uri=CELEX:32017R0745 tiende a incorporar un número significativamente mayor de productos dentro del ámbito sanitario, incluyendo algunos softwares y wearables con finalidades preventivas. Paralelamente, el AI Act[1] https://eur-lex.europa.eu/eli/reg/2024/1689/oj/eng?utm_source=chatgpt.com introduce un enfoque de gestión del riesgo para sistemas de inteligencia artificial en salud, aunque sin resolver aún la frontera entre bienestar y medicina (6).
Por su parte, Australia (TGA) y Singapur (HSA) han adoptado enfoques más próximos al estadounidense, mientras que Reino Unido (MHRA) se encuentra en pleno proceso de reforma para simplificar la regulación de software médico. La convergencia internacional en torno al enfoque basado en uso previsto sugiere que la guía de la FDA puede actuar como referencia de facto para otros mercados.
Implicancias para telemedicina y ecosistema sanitario latinoamericano. Si bien en América Latina la regulación sanitaria de la salud digital aún se encuentra en proceso de consolidación, el mercado de dispositivos orientados a bienestar y auto-seguimiento ha experimentado una expansión acelerada, particularmente en segmentos vinculados al deporte, la computación vestible (wearable computing) y la cuantificación personal (self-tracking).
Para agencias regulatorias de sólida trayectoria como la ANMAT (Argentina), la ANVISA (Brasil) y el INVIMA (Colombia), la adopción de un marco de delimitación funcional permitiría evitar la medicalización regulatoria[2] de productos de consumo, liberar capacidad institucional hacia tecnologías de mayor riesgo sanitario, favorecer la convergencia regulatoria con mercados de exportación y reducir la incertidumbre normativa para la industria tecnológica.
De manera indirecta, la guía también habilita esquemas de telemonitorización no clínica, en los cuales el usuario registra variables orientadas a bienestar sin que ello genere obligación de evaluación o intervención sanitaria por parte del sistema de salud.
Conclusión
La actualización de la Guía General Wellness Policy for Low Risk Devices delimita con mayor precisión el ámbito no sanitario del bienestar digital bajo una lógica funcional. Al adoptar el uso previsto y el riesgo como criterios centrales, la FDA proporciona una herramienta para ordenar el ecosistema digital y reducir la incertidumbre regulatoria. La guía puede influir en la formación de estándares internacionales y resulta relevante para jurisdicciones que buscan equilibrar innovación tecnológica con protección sanitaria, especialmente en contextos de telemedicina y digitalización del cuidado.
Referencias
- U.S. Food and Drug Administration. General Wellness: Policy for Low Risk Devices. Silver Spring (MD): FDA; 2026 Jan 6. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/general-wellness-policy-low-risk-devices
- U.S. Food and Drug Administration. Digital Health Center of Excellence. Silver Spring (MD): FDA; 2020. https://www.fda.gov/medical-devices/digital-health-center-excellence
- United States Congress. 21st Century Cures Act. Pub. L. 114-255; 2016 Dec 13. https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act
- U.S. Food and Drug Administration. Policy for Device Software Functions and Mobile Medical Applications. Silver Spring (MD): FDA; 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-device-software-functions-and-mobile-medical-applications
- U.S. Food and Drug Administration. Clinical Decision Support Software Guidance. Silver Spring (MD): FDA; 2024. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/clinical-decision-support-software
- European Parliament and the Council of the European Union. Artificial Intelligence Act. Brussels; 2025. https://artificialintelligenceact.eu/
(*) Autora. María Isabel IÑIGO PETRALANDA MBA, MBE www.linkedin.com/in/maria-isabel-inigo-petralanda , es abogada, Bioeticista, especialista en dominios éticos jurídicos y operativos de tecnologías data driven e informática sanitaria desde etapas de investigación hasta su despliegue clínico. Es DCO para salud digital. Es consultora de Gobiernos y empresas, integra comités de ética en investigación y es docente de universidades en argentina y el extranjero. Orgullosa colaboradora de contenido editorial del medio https://saludenlinea.press/
[1] El AI Act es el primer marco legal sobre IA de la UE que aborda los riesgos de los sistemas de IA y promueve un desarrollo y despliegue responsables.”
[2] A lo que en este trabajo denomino “medicalización regulatoria”, entendido como over-classification de bienes de consumo bajo categorías sanitarias propias de productos médicos, fenómeno tratado por FDA y EMA como “device boundary”o “regulatory overreach”.